Nobel de química 2024 por la predicción de la estructura de proteínas
Ninfa Jiménez Acosta, Juan Pablo Martínez Sáenz y Abraham Vidal Limón*
Este año 2024 ha sido muy excitante para la química de proteínas y las ciencias computacionales debido a que el Premio Nobel en Química 2024 ha sido otorgado a David Baker (Washington, 1962), director del Institute for Protein Design de la Universidad de Washington, Demis Hassabis (Londres, 1976) y John Michael Jumper (Arkansas, 1985), cofundadores de la empresa DeepMind por las contribuciones derivadas de sus estudios computacionales sobre la estructura de proteínas.
Baker es la mente maestra detrás de RoseTTAFold (2021), un programa que es capaz de resolver la complicada estructura tridimensional que adopta una proteína en el espacio (lo que denominamos ¨plegamiento¨) y que le permite tener alguna función. Además, este programa desarrollado en su laboratorio permite diseñar proteínas desde cero, con funciones novedosas (Figura 1).

Fig 1. Ganadores del Premio Nobel de Química 2024 por el diseño computacional de proteínas y la predicción de su estructura. The Nobel Prize in Chemistry 2024. NobelPrize.org. Nobel Prize Outreach AB 2024. Thu. 24 Oct 2024. <https://www.nobelprize.org/prizes/chemistry/2024/summary/>
Por otro lado, Hassabis y Jumper han contribuido significativamente para que el uso de inteligencia artificial (IA) y, particularmente el aprendizaje profundo, sea una herramienta que facilite la predicción de la estructura tridimensional de las proteínas. AlphaFold es un sistema de IA que funciona de esta forma, haciendo la predicción a partir de sus secuencias de aminoácidos (moléculas fundamentales que conforman a las proteínas) y, ya que no sólo sirve para conocer las posiciones atómicas, sino que abre la puerta a la comunidad científica a poder modelar las interacciones entre las proteínas, impulsando la investigación en el área de desarrollo de fármacos. Ambos esfuerzos computacionales han permitido entender procesos bioquímicos en situaciones donde los métodos experimentales no han podido, ayudando a revelar los secretos de las proteínas y sus funciones.
El comité del Karolinska Institutet (Suecia), órgano encargado de asignar este galardón, ha resaltado la importancia de estos logros científicos en diversas áreas del conocimiento. Desde el desarrollo acelerado de vacunas, la generación de nuevos bionanomateriales, pasando por el diseño de fármacos dirigidos para tratar el cáncer hasta sus usos en la industria de la química verde. Heiner Linke, presidente del comité Karolinska declaró que: “Uno de los descubrimientos de este año se refiere a la construcción de proteínas espectaculares. El otro consiste en hacer realidad un sueño de 50 años: predecir las estructuras de las proteínas a partir de sus secuencias de aminoácidos. Ambos descubrimientos abren enormes posibilidades”.
El diseño de proteínas y sus múltiples usos
Las proteínas están conformadas, generalmente, por 20 tipos de aminoácidos que representan los bloques básicos de la vida. Desde 2003, Baker y su grupo de investigación trabajaron en utilizar estos bloques para diseñar proteínas nuevas que no se parecieran a ninguna otra. Desde entonces, su interés principal se ha volcado en el diseño de una proteína tras otra, incluidas aquellas con usos en las áreas de vacunas, fármacos, biosensores y bionanomateriales, con ayuda de RoseTTAFold.
De manera paralela, Jumper y Hassabis también enfocaron su investigación en la predicción de estructuras proteicas. Si bien en las proteínas los aminoácidos están unidos en largas cadenas que se pliegan para formar una estructura tridimensional, las posibles soluciones al acomodo de los átomos de los aminoácidos en el espacio son muchas y definen la actividad y función de las proteínas. Es así como encontramos proteínas que comparten algunas secuencias similares en las uñas, el cabello y los vasos sanguíneos, pero cuyo arreglo tridimensional es distinto. Desde hace más de 50 años diversos grupos de investigación han intentado predecir las estructuras de las proteínas a partir de secuencias de aminoácidos, sin embargo, esta tarea no es fácil. Hasta hace cuatro años se produjo un avance asombroso con el uso de recursos computacionales de Google y que derivaron en la compañía DeepMind. En ese entonces, Hassabis y Jumper nombraron a su modelo de IA AlphaFold2 y, con su ayuda, han podido predecir la estructura de millones de proteínas. Sobra decir que, desde su liberación, el programa AlphaFold2 ha sido utilizado por más de dos millones de usuarios alrededor del mundo, lo cual deja en claro su impacto en la sociedad en diversas áreas. Dicho impacto y los avances en la predicción y el diseño de estructuras de proteínas representan un área de múltiples oportunidades, donde destaca la medicina de precisión, en la que sería posible diseñar fármacos que actúen de manera específica en el tiempo y tejidos del organismo.
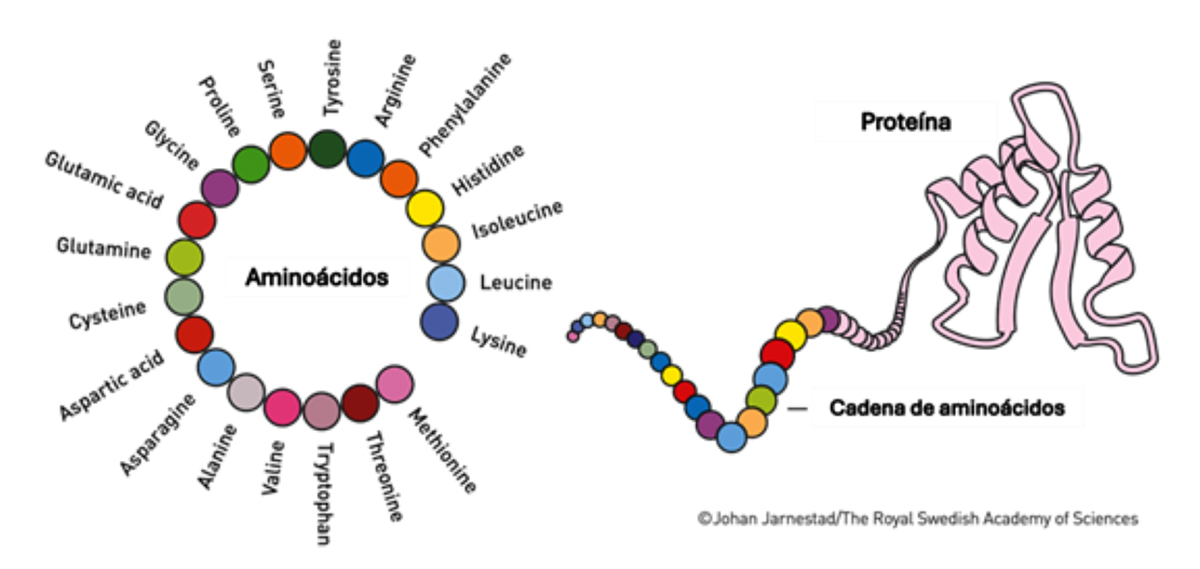
Fig 2. Estructura de las proteínas, que están conformadas por moléculas denominadas aminoácidos
¿Cómo usamos estas aplicaciones en el INECOL?
De esta manera, en el Instituto de Ecología A.C. (INECOL) trabajamos con herramientas de IA que pueden ayudarnos a predecir cómo una molécula (por ejemplo, un posible fármaco) interacciona con otra molécula objetivo (receptor) en las células y así tratar alguna enfermedad. Otra de sus funciones puede ser diseñar nuevos compuestos (aminoácidos, péptidos o proteínas) que sean parecidos a los que ya sabemos que funcionan, haciendo una comparación que nos ayude a descubrir posibles tratamientos para enfermedades, por ejemplo, la diabetes. Pero ¿cómo se logra esto? y ¿qué papel tiene la inteligencia artificial?
El aprendizaje automático es una forma de inteligencia artificial que nos permite clasificar objetos por medio de sus características. En nuestro caso, los objetos pueden ser moléculas y, las características que queremos comparar, sus propiedades químicas y físicas. Así, por medio de diversos métodos (algoritmos) de clasificación, podemos comparar moléculas de compuestos extraídos de plantas, insectos o diversos productos naturales con los que ya sabemos que funcionan, filtrándolos para disminuir la cantidad de objetos de trabajo y pasando a otros métodos computacionales (como la simulación molecular), para ahorrar tiempo y dinero para el posterior trabajo experimental.
Estamos en una época de gran desarrollo tecnológico y tenemos la capacidad de aprovechar al máximo las herramientas computacionales para resolver problemas en química, bioquímica y biología de una manera novedosa. La investigación de Baker, Jumper y Hassabis abre la puerta para explorar nuevas fronteras en la ciencia y comprender diversos procesos biológicos.

Fig 3. Herramientas computacionales utilizadas para el análisis y predicción de proteínas (Abraham Vidal-Limon, José E. Aguilar-Toalá, and Andrea M. Liceaga. Journal of Agricultural and Food Chemistry 2022 70 (4), 934-943
Referencias
- https://saluddigital.com/es/big-data/cientificos-de-google-obtienen-premio-nobel-de-quimica-por-su-trabajo-en-alphafold/
- The Nobel Prize in Chemistry 2024. https://www.nobelprize.org/prizes/chemistry/2024/summary/